This post will recap the previous,
Haskell Meets the Central Dogma,
with the intent to introduce algebraic data
types.
Using these will allow refinement of the design, present some more
features of Haskell, and continue a gradual exposition.
The first in this series, A Review of the Central Dogma,
reviews the biological context of the functions presented.
I previously began by declaring some type synonyms in my code so that I could
then differentiate between functions meant to operate with DNA (DNASeq),
RNA (RNASeq), or protein (ProteinSeq). The type synonyms were all equivalent
to Haskell’s String type: [Char]. Here is that code for quick reference:
typeElement=Char-- A unitary objecttypeSeq=[Element]-- An ordered collection of ElementstypeDNABase=Element-- DNA variant of ElementtypeDNASeq=[DNABase]-- DNA variant of SequencetypeRNABase=Element-- RNA variant of ElementtypeRNASeq=[RNABase]-- RNA variant of SequencetypeAminoAcid=Element-- Protein variant of ElementtypeProteinSeq=[AminoAcid]-- Protein variant of Sequence
With these in place I could declare translate :: RNASeq -> ProteinSeq in lieu
of translate :: String -> String to indicate that the expected behavior is
meaningful for particular Strings values, not String values in general. Yet
since these declarations are fundamentally the same, we are still able to do
some weird things that it might be best to prevent:
ghci>letmydna="GATTACA"::DNASeqghci>letmypro="ALGLYSW"::ProteinSeqghci>mydna++mypro-- we can concatenate, but what kind of molecule is that?!"GATTACAALGYLSW"ghci>:tmydna++mypro-- result type of concatenation in ghci?mydna++mypro::[DNABase]ghci>:tmypro++mydna-- it depends on the order!mypro++mydna::[AminoAcid]ghci>:treverseTranslatereverseTranslate::ProteinSeq->[[DNACodon]]ghci>reverseTranslatemydna-- The base Chars in DNA can interpreted as valid amino acids[["GGA","GGC","GGG","GGT"],["GCA","GCC","GCG","GCT"],["ACA","ACC","ACG","ACT"],["ACA","ACC","ACG","ACT"],["GCA","GCC","GCG","GCT"],["TGC","TGT"],["GCA","GCC","GCG","GCT"]]
The takeaway lesson here is that while using type to create synonyms can help
with code readability where you’d like to tersely annotate something with a
comment (it’s like laying a comment on top of your type), they don’t
meaningfully change anything below the surface.
Let’s explore using some algebraic data types to represent our
data which will simultaneously improve the performance, clarity, and rigor of
the code.
Algebraic types with data
So let’s begin by intoducing some custom data types to represent nucleic acid
sequences and protein sequences. I will not preserve the conceptual distinction
between DNA and RNA at the type level as I did before; because they are
directly correlated (to the extent that we are concerned here), it makes sense
to think of DNA and RNA as simply different representations of the same
underlying nucleic acid sequence. This is a common convention in
bioinformatics, and it will help to keep reduce the amount and complexity of our
code. I will not cover ambiguous nucleotide or amino acids here.
--A Nucleotide may be either Adenine, Cytosine, Guanosine, or Thymine/Uracil--Thymine is Uracil in RNAdataNucleotide=Adenine|Cytosine|Guanosine|Thyminederiving(Eq,Ord,Show)--An AminoAcid may be one of the 20 standard amino acids, or a StopdataAminoAcid=Alanine|Arginine|Asparagine|Aspartate|Cysteine|Glutamate|Glutamine|Glycine|Histidine|Isoleucine|Leucine|Lysine|Methionine|Phenylalanine|Proline|Serine|Threonine|Tryptophan|Tyrosine|Valine|Stopderiving(Eq,Show)typeNucleicSeq=[Nucleotide]-- Nucleic Acid SequencetypeProteinSeq=[AminoAcid]-- Protein Sequence
Here I’ve created two new data types: Nucleotide, and AminoAcid. These new
data types have their own unique value constructors. The type synonyms
NucleicSeq and ProteinSeq suggest my intent to work with these sequences as
lists of their respective subunits.
Let’s get a quick feel for this system:
ghci>letmydna=[Guanosine,Adenine,Thymine,Thymine,Adenine,Cytosine,Adenine]ghci>letmypro=[Alanine,Leucine,Glycine,Tyrosine,Leucine,Serine,Tryptophan]ghci>showmydna"[Guanosine,Adenine,Thymine,Thymine,Adenine,Cytosine,Adenine]"ghci>showmypro"[Alanine,Leucine,Glycine,Tyrosine,Leucine,Serine,Tryptophan]"ghci>mydna++mypro-- Lists are homogenous, we can't mix types in a sequence-- Couldn't match type ‘AminoAcid’ with ‘Nucleotide’-- Expected type: [Nucleotide]-- Actual type: [AminoAcid]-- In the second argument of ‘(++)’, namely ‘mypro’-- In the expression: mydna ++ myproghci>Thymine:mydna-- Prepend Thymine[Thymine,Guanosine,Adenine,Thymine,Thymine,Adenine,Cytosine,Adenine]
Because lists in Haskell are homogenous structures, we are now sure that we
cannot compile some code in which we’ve accidentally tried to combine the types
into one value. Since the types are distinct, it is now clear to both the reader
and the compiler that translate :: NucleicSeq -> ProteinSeq uses
[Nucleotide] as its input type.
It looks like working with this system could get a little cumbersome in terms of
typing: "GATTACA" was much easier to type than [Guanosine, Adenine, Thymine,
Thymine, Adenine, Cytosine, Adenine]. Let’s take a moment to write some helpful
functions for nicely inputting to and outputting from this model:
charToNuc::Char->NucleotidecharToNuc'A'=AdeninecharToNuc'C'=CytosinecharToNuc'G'=GuanosinecharToNuc'T'=ThyminecharToNuc'U'=ThyminecharToNucx=error$x:" is not a valid nucleotide character"stringToNucleicSeq::String->NucleicSeqstringToNucleicSeq=mapcharToNucdnaSymbol::Nucleotide->ChardnaSymbolAdenine='A'dnaSymbolCytosine='C'dnaSymbolGuanosine='G'dnaSymbolThymine='T'rnaSymbol::Nucleotide->CharrnaSymbolThymine='U'rnaSymbolx=dnaSymbolxrepresentAsDNA::NucleicSeq->StringrepresentAsDNA=mapdnaSymbolrepresentAsRNA::NucleicSeq->StringrepresentAsRNA=maprnaSymbolaaSymbolTable=[(Alanine,'A'),(Arginine,'R'),(Asparagine,'N'),(Aspartate,'D'),(Cysteine,'C'),(Glutamate,'E'),(Glutamine,'Q'),(Glycine,'G'),(Histidine,'H'),(Isoleucine,'I'),(Leucine,'L'),(Lysine,'K'),(Methionine,'M'),(Phenylalanine,'F'),(Proline,'P'),(Serine,'S'),(Threonine,'T'),(Tryptophan,'W'),(Tyrosine,'Y'),(Valine,'V'),(Stop,'*')]representProtein::ProteinSeq->StringrepresentProtein=mapfetchAAwherefetchAAx=fromJust$lookupxaaSymbolTablestringToProteinSeq::String->ProteinSeqstringToProteinSeq=mapfetchSymbolwherefetchSymbolx=fromMaybe(error("Invalid amino acid: "++showx))$lookupx$invertaaSymbolTableinvert=mapswapswap(x,y)=(y,x)
So now where it is more convenient we have the ability to make sequences from
Strings and to produce Strings from our sequences like so:
I should note that performing lookups on lists like I have done here is not the
most efficient method. Data structures such as Map or HashMap will
have better lookup performance.
Re-implementation of Complementation
With this new data model in place, let’s implement the central dogma functions
agin. Since replication is so trivial (I’ll say a little more on this at the
end), I’ll move right into complementation. This is exactly the same
as before but with Nucleotide values instead of Char values.
complementNucleotide expresses the Nucleotide-wise rules of complementation
and complement maps it over a list to generate the complementary list.
reverseComplement uses function composition to create a function that returns
the reverse result of the complement function (putting it into the standard 5’
to 3’ orientation). Put in practice:
ghci>complement[Thymine,Adenine,Cytosine,Guanosine][Adenine,Thymine,Guanosine,Cytosine]ghci>complement$stringToNucleicSeq"GATTACA"[Cytosine,Thymine,Adenine,Adenine,Thymine,Guanosine,Thymine]ghci>reverseComplement$stringToNucleicSeq"GATTACA"[Thymine,Guanosine,Thymine,Adenine,Adenine,Thymine,Cytosine]ghci>reverseComplement[Glutamate]-- Disallowed-- Couldn't match expected type ‘Nucleotide’-- with actual type ‘AminoAcid’-- In the expression: Glutamate-- In the first argument of ‘reverseComplement’, namely ‘[Glutamate]’
Translation
The previous implementation for the geneticCode function was written to
pattern match on strings of three characters which representing a codon. If the
function was given a string of a different length and/or a string containing an
invalid character, it would produce '-' to denote that it is not meaningful.
Here is the first two lines of my previous definition of geneticCode:
In the former there is no guarantee whether the characters in the input
are valid while in the latter we have that assurance. Yet in both functions
there is the possibility for lists of incorrect length; I would like to fix that
so I will formalize a codon data type and write the function to fit.
Now I have a new data type called Codon whose values can be created using the
type constructor Codon (same name in this case, but it’s good to remember the
distinction between types and their constructors) which takes three and only
three Nucleotide values and holds them as an ordered triplet. I have prepended
each field with ! to make them strict, essentially establishing a policy that
a Codon cannot be partially defined.
This provides the basis of the new definition of geneticCode:
Thanks to some help from vim, I didn’t have to invest much time into retyping
that. Now to create the new translate function:
--Convert a [Nucleotide] to [Codon], trimming possible remainderasCodons::NucleicSeq->[Codon]asCodonss=mapMaybetoCodon(chunksOf3s)wheretoCodon[i,j,k]=Just(Codonijk)toCodon_=Nothingtranslate::NucleicSeq->ProteinSeqtranslates=mapgeneticCode$asCodonss
translate :: NucleicSeq -> ProteinSeq makes use of asCodons to convert the
nucleotides to codons and maps geneticCode over the result. asCodons does
the conversion by chunking the input into triplet lists whose values are then
used to construct a Codon in toCodon. If toCodon gets a list of length
other than 3, because the number of input Nucleotide values was an integer
multiple of 3, it produces a Nothing. The mapMaybe in asCodons discards
Nothing values and extracts the values from the Just.
The first key to the puzzle of computing reverse translation will be a
function that maps each AminoAcid to its corresponding list of coding
Codons. I’ll also define a convenience function mkCodon (“make Codon”) to
help keep the lines a little shorter.
mkCodon::String->CodonmkCodon[f,s,t]=Codon(charToNucf)(charToNucs)(charToNuct)mkCodon_=error"Incorrect number of characters to make Codon"codonsFor::AminoAcid->[Codon]codonsForAlanine=[mkCodon"GCA",mkCodon"GCC",mkCodon"GCG",mkCodon"GCT"]codonsForCysteine=[mkCodon"TGC",mkCodon"TGT"]codonsForAspartate=[mkCodon"GAC",mkCodon"GAT"]codonsForGlutamate=[mkCodon"GAA",mkCodon"GAG"]codonsForPhenylalanine=[mkCodon"TTC",mkCodon"TTT"]codonsForGlycine=[mkCodon"GGA",mkCodon"GGC",mkCodon"GGG",mkCodon"GGT"]codonsForHistidine=[mkCodon"CAC",mkCodon"CAT"]codonsForIsoleucine=[mkCodon"ATA",mkCodon"ATC",mkCodon"ATT"]codonsForLysine=[mkCodon"AAA",mkCodon"AAG"]codonsForLeucine=[mkCodon"CTA",mkCodon"CTC",mkCodon"CTG",mkCodon"CTT",mkCodon"TTA",mkCodon"TTG"]codonsForMethionine=[mkCodon"ATG"]codonsForAsparagine=[mkCodon"AAC",mkCodon"AAT"]codonsForProline=[mkCodon"CCA",mkCodon"CCC",mkCodon"CCG",mkCodon"CCT"]codonsForGlutamine=[mkCodon"CAA",mkCodon"CAG"]codonsForArginine=[mkCodon"AGA",mkCodon"AGG",mkCodon"CGA",mkCodon"CGC",mkCodon"CGG",mkCodon"CGT"]codonsForSerine=[mkCodon"AGC",mkCodon"AGT",mkCodon"TCA",mkCodon"TCC",mkCodon"TCG",mkCodon"TCT"]codonsForThreonine=[mkCodon"ACA",mkCodon"ACC",mkCodon"ACG",mkCodon"ACT"]codonsForValine=[mkCodon"GTA",mkCodon"GTC",mkCodon"GTG",mkCodon"GTT"]codonsForTryptophan=[mkCodon"TGG"]codonsForTyrosine=[mkCodon"TAC",mkCodon"TAT"]codonsForStop=[mkCodon"TAA",mkCodon"TAG",mkCodon"TGA"]
The reverse translation operation can just be the application of codonsFor to
each AminoAcid in a ProteinSeq:
Now let’s create uniqueCodings :: ProteinSeq -> [NucleicSeq] which will
produce all of the possible nucleic acid sequences which could code for the
protein. I’ll have to add an intermediate step to unpack the Nucleotide
components from a Codon.
Beware: the size of the output increases exponentially with the
size of the input (unless you only have Methionine or Tryptophan in the
sequence).
-- Unpack the Codons in [Codon] to get [NucleicSeq]unCodons::[Codon]->[NucleicSeq]unCodons=mapunCodonwhereunCodon(Codonfst)=[f,s,t]--produce all the ways the protein sequence could be encodeduniqueCodings::ProteinSeq->[NucleicSeq]uniqueCodingss=foldr(liftA2(++).unCodons)([[]]::[NucleicSeq])$reverseTranslates--Count up all the ways the protein sequence could be encodeduniqueCodingsCount::ProteinSeq->IntegeruniqueCodingsCounts=fold((*).genericLength)1$reverseTranslates
Note that I have modified uniqueCodings to make use of the lists as
applicative functors. liftA2 is from Control.Applicative. You may want to
start reading here
if applicative functors are new to you.
With the appropriate adjustment to constrainedUniqueCodings I can repeat my
previous work on finding out whether a certain NucleicSeq can be found among
the reverse translated NucleicSeqs from a given ProteinSeq.
Fidelitous (error-free) replication is such a trivial problem that we needn’t
hardly think about it. The function which always gives back the value it was
given is known as the identity function.
The replication of a sequence can then be expressed by using the Haskell
identity function id, which is:
id::a->aidx=x
So if we cared to, an alias for id could be made like so:
replication=id
The story of replication probably won’t get interesting again until we consider
infidelity (scandalous!).
This post will introduce some Haskell code to perform sequence operations
relevant to the central dogma of molecular biology. The code here should be
relatively simple, but some slight familiarity with Haskell or functional programming
is assumed.
The treatment of biology in this post is also kept simple, though if you don’t know
what the central dogma of molecular biology is or could do with a brief
review, I have written a bit on it;
it is meant to give a little background for some of the molecular biology terms
used in this post, and to describe its context in the scope of biology today.
I’ll be working with sequences here using Haskell’s native String type. Though
DNA, RNA, and protein sequences will all share this same underlying type it will
be helpful, in terms of documenting the code, to be able to convey whether a
function is meant to work with a particular kind of sequence or another. To do
this let’s make use of the type declaration which creates type synonyms.
These should help clarify the code’s intent.
Note that in Haskell a string is a special case of a list, [], one that contains Char elements:
[Char]. These will be more informative than just Char and String later on:
typeElement=Char-- A unitary objecttypeSeq=[Element]-- An ordered collection of ElementstypeDNABase=Element-- DNA variant of ElementtypeDNASeq=[DNABase]-- DNA variant of SequencetypeRNABase=Element-- RNA variant of ElementtypeRNASeq=[RNABase]-- RNA variant of SequencetypeAminoAcid=Element-- Protein variant of ElementtypeProteinSeq=[AminoAcid]-- Protein variant of Sequence
Replication, Replication
So let’s get started with replication, one of the basic operations of DNA and RNA
in which an exact copy (in its own medium) is produced. Since replication (when
we ignore the possibility of error) can be modeled by defining a function that
simply returns its own input, it’s rather trivial to implement. Here I’ll choose
to define a function that works element-wise, and a sequence-wise function which
is the mapping of the element-wise one over all of its elements. This pattern will be
employed throughout.
--function that gives whatever sequence element it receivesreplicateBase::Element->ElementreplicateBasex=x--function that maps the replicateBase function over its inputreplication::Seq->Seqreplications=mapreplicateBases
When we supply a sequence string to the replication function like
replication "GATTACA", it should give us "GATTACA" when it evaluates.
You could try this out with various sequence strings, the empty string is an
important test as well. Here’s a little function to help test if the results are good.
--True if the result of the replication function is the same as the inputvalidReplication::Seq->BoolvalidReplications=s==replications
Everybody likes complements
Now let’s take a look at how we can write code to get the complementary sequence
for a DNA or RNA sequence. Recall that DNA and RNA form double stranded
structures according to nucleobase complementarity: A pairs with T (or U
in RNA) and C pairs with G. Here’s a representation of this in code:
complementDNABase::DNABase->DNABasecomplementDNABase'A'='T'complementDNABase'C'='G'complementDNABase'G'='C'complementDNABase'T'='A'complementDNABasex=x-- do not change unexpected characterscomplementDNA::DNASeq->DNASeqcomplementDNAs=mapcomplementDNABasescomplementRNABase::RNABase->RNABasecomplementRNABase'A'='U'complementRNABase'C'='G'complementRNABase'G'='C'complementRNABase'U'='A'complementRNABasex=x-- do not change unexpected characterscomplementRNA::RNASeq->RNASeqcomplementRNAs=mapcomplementRNABases
These examples leverage pattern matching on their input. Note that we also have
the option of writing it with guards, though I don’t prefer guard syntax for pattern
matching. Here’s complementDNABase using guards just to demonstrate:
Giving "GATTACA"to complementDNA should yield "CTAATGT" and giving the
corresponding "GAUUACA" to complementRNA should yield "CUAAUCU".
Complementation is a reciprocal process, meaning that if we produce s' as the
complement of s, then s is also the complement of s'. This is an important
property of the function, so let’s test it by making sure that if either of the
functions reproduce the input when executed twice in succession.
Use these functions on any sequence string to see if the function was correctly
written.
DNA and RNA sequences are traditionally presented in a specific direction: 5’ to
3’ (details here).
It is important to remember that the complementing strand of DNA or RNA
generally “runs” in the opposite direction. That is to say, if you take GATTACA
which is reading 5’ to 3’, and produce it’s complement CTAATGT, the complement
produced is reading 3’ to 5’. It is common then to refer to the “reverse
complement” of a sequence as the complement read in the conventional 5’ to 3’
fashion. Producing such a reverse complement function in Haskell is simple:
Here I’ve used function composition with Haskell’s
reverse
function to easily define the reverse complement functions. That’s pretty handy.
Transcription and noitpircsnarT
The process of transcription refers to the production of RNA from a DNA template
and reverse transcription the production of DNA from an RNA template. This is
again mediated by base complementarity, and the code for it can be readily made
by adapting our complementing functions from above. When a DNA sequence, s, is
transcribed, a copy of that sequence is made in RNA using base complementarity
to the complementing DNA strand, s'. Since RNA nucleobases differ in the use
of Uracil instead of Thymine, the matter of transcription involves replacing
our 'T's with 'U's and vice versa for reverse transcription.
transcribeBase::DNABase->RNABasetranscribeBase'T'='U'transcribeBasex=x-- leave other bases, or unexpected chars, alonetranscribe::DNASeq->RNASeqtranscribes=maptranscribeBasesreverseTranscribeBase::RNABase->DNABasereverseTranscribeBase'U'='T'reverseTranscribeBasex=x-- leave other bases, or unexpected chars, alonereverseTranscribe::RNASeq->DNASeqreverseTranscribes=mapreverseTranscribeBases
Transcription and reverse transcription functions should be symmetrical. By
which I mean to say that if one transcribes a DNA sequence and then reverse
transcribes the result, it should reproduce the input. The same is true for
reverse transcription of an RNA sequence then transcription of the result. We can
make some fun functions to test whether this holds true:
These only really work if the inputs are valid. Passing it a mixed
sequence like “GATUACA” should evaluate False in both functions.
Translation -> Traducción
Translation is the process of creating proteins from messenger RNA sequences.
RNA is read in chunks of three nucleotides, which constitute a codon, to
determine which amino acid to add to a growing protein chain. The “Genetic Code”
is the (mostly universal) coding scheme used in biology that maps codons to
amino acids. Without further ado, here’s the genetic code in haskell:
--Codons are just special cases (length 3) of their basic typestypeDNACodon=DNASeqtypeRNACodon=RNASeq--Using RNA codons, if we want to use DNA, we can just transcribe first--There are 4^3 = 64 combinations, so 64 codons. geneticCode::RNACodon->AminoAcidgeneticCode"AAA"='K'-- LysinegeneticCode"AAC"='N'-- AsparaginegeneticCode"AAG"='K'-- LysinegeneticCode"AAU"='N'-- AsparaginegeneticCode"ACA"='T'-- ThreoninegeneticCode"ACC"='T'-- ThreoninegeneticCode"ACG"='T'-- ThreoninegeneticCode"ACU"='T'-- ThreoninegeneticCode"AGA"='R'-- ArgininegeneticCode"AGC"='S'-- SerinegeneticCode"AGG"='R'-- ArgininegeneticCode"AGU"='S'-- SerinegeneticCode"AUA"='I'-- IsoleucinegeneticCode"AUC"='I'-- IsoleucinegeneticCode"AUG"='M'-- MethioninegeneticCode"AUU"='I'-- IsoleucinegeneticCode"CAA"='Q'-- GlutaminegeneticCode"CAC"='H'-- HistidinegeneticCode"CAG"='Q'-- GlutaminegeneticCode"CAU"='H'-- HistidinegeneticCode"CCA"='P'-- ProlinegeneticCode"CCC"='P'-- ProlinegeneticCode"CCG"='P'-- ProlinegeneticCode"CCU"='P'-- ProlinegeneticCode"CGA"='R'-- ArgininegeneticCode"CGC"='R'-- ArgininegeneticCode"CGG"='R'-- ArgininegeneticCode"CGU"='R'-- ArgininegeneticCode"CUA"='L'-- LeucinegeneticCode"CUC"='L'-- LeucinegeneticCode"CUG"='L'-- LeucinegeneticCode"CUU"='L'-- LeucinegeneticCode"GAA"='E'-- GlutamategeneticCode"GAC"='D'-- AspartategeneticCode"GAG"='E'-- GlutamategeneticCode"GAU"='D'-- AspartategeneticCode"GCA"='A'-- AlaninegeneticCode"GCC"='A'-- AlaninegeneticCode"GCG"='A'-- AlaninegeneticCode"GCU"='A'-- AlaninegeneticCode"GGA"='G'-- GlycinegeneticCode"GGC"='G'-- GlycinegeneticCode"GGG"='G'-- GlycinegeneticCode"GGU"='G'-- GlycinegeneticCode"GUA"='V'-- ValinegeneticCode"GUC"='V'-- ValinegeneticCode"GUG"='V'-- ValinegeneticCode"GUU"='V'-- ValinegeneticCode"UAA"='*'-- Stop (Ochre)geneticCode"UAC"='Y'-- TyrosinegeneticCode"UAG"='*'-- Stop (Amber)geneticCode"UAU"='Y'-- TyrosinegeneticCode"UCA"='S'-- SerinegeneticCode"UCC"='S'-- SerinegeneticCode"UCG"='S'-- SerinegeneticCode"UCU"='S'-- SerinegeneticCode"UGA"='*'-- Stop (Opal)geneticCode"UGC"='C'-- CysteinegeneticCode"UGG"='W'-- TryptophangeneticCode"UGU"='C'-- CysteinegeneticCode"UUA"='L'-- LeucinegeneticCode"UUC"='F'-- PhenylalaninegeneticCode"UUG"='L'-- LeucinegeneticCode"UUU"='F'-- PhenylalaninegeneticCodex='-'-- Anything else corresponds to nothing
When it comes to expressing coding schemes, there’s just no way to cut corners
so that got a little verbose. Writing that sort of code is no fun, but there was
no way around it1. So now that we’ve got our genetic code, let’s put it
to work translating some codons to a protein sequence.
chunksOf 3 creates a function that takes a list, and returns a list of lists
of length 3, though the last item might be shorter if the input list is not evenly
divisible by 3. We can feed our sequence string to this, strings are just lists
of characters after all, and get a list of codons back. chunksOf 3 "GAUUACA"
would produce ["GAU", "UAC", "A"]. Mapping the genetic code function over that
list of strings would give "DY-".
If we want to translate DNA sequences, we
could go the hard way and write a new function, or we could go the smart way and
create easily with function composition:
The idea now is to make sure that the results of our translation function when
fed the mRNA sequence jives with the reference implementation’s results. Let’s
get down to work! I don’t want to have to type or copy the sequence into the
source, so let’s make a function to read the sequences out of the files.
--A very simple, non-general function to get the sequencesimpleSeqGetter::FilePath->IOSeqsimpleSeqGetterfp=dounmunged<-readFilefpletlinedropped=drop1$linesunmungedreturn$foldr(++)""linedropped
This is not a proper FASTA parsing job, but it serves for now. I’ve dropped the
first line describing the sequence and gotten rid of the newline characters. So
now I can do the following in ghci:
It’s pretty clear that our results don’t match. Some readers might have seen
this coming, because I’ve not prepared to handle an important detail! If you
look carefully you’ll see that the expected sequence is there, but sandwiched
between some other protein sequence. Without getting into specifics, mRNA sequences
contain untranslated regions (UTRs) at both the 5’ and 3’
ends of the sequence. These untranslated regions are a part of the sequence file;
to figure out what the ribosome would actually produce from this particular
mRNA we’ll need to know how to drop them (UTRs are totally interesting in their
own right, but that’s a story for another time).
To determine where the translated section begins, we must look for the first (5’
most) AUG codon, this is the start codon and codes for Methionine.
Everything from that point on is
translated until we reach a stop codon: one of UAA, UAG, UGA. The region
from the start codon to the stop codon is the coding region.
Let’s implement these rules in code now:
This now drops everything before the start codon, and keeps everything until the
stop codon. Now we can try this function out and test its results in ghci:
Tada! We can translate a given DNA or RNA sequence as well as fish out the first coding
region from a sequence. By the way, have you thought it’s weird that I’ve been
using translateDNA on an RNA sequence? Yeah, it’s not just you. Nucleic acid
sequences are often stored in the DNA alphabet, regardless of whether it makes
more physical/biological sense as RNA. It’s a bit odd, but common enough to
gloss over transcription and reverse transcription since they directly correlate.
Translation Party: Reverse Translation
In my previous post,
I made something of a big deal about how reverse
translation (also sometimes called “back translation”)
has never been observed and we’ve got pretty good reasons to never
expect it to occur. But it turns out that there are plenty of times where we
might want to address reverse translation computationally.
Most amino acids have more than one codon that codes for them, but no codon
codes for more than one amino acid. In this way, the genetic code is degenerate
but not ambiguous. In other words, a given nucleic acid sequence passed through
the genetic code will only produce one protein sequence, but a given protein
sequence could be decoded to multiple nucleic acid sequences.
This leads to a very hard problem in trying to represent all the possible
encoding sequences. Only for rather short protein sequences is it computationally
feasible to consider representing all the combinations in memory or storage, or
to iteratively consider all of the possibilities.
I think that looking deeper into reverse translation applications is worthy of
one or more focused posts, but it’d be good to give a short treatment here. I’ll
define a function that decodes a protein sequence to its possible DNA sequences.
Might get a little lengthy again…
codonsFor::AminoAcid->[DNACodon]codonsFor'A'=["GCA","GCC","GCG","GCT"]-- AlaninecodonsFor'C'=["TGC","TGT"]-- CysteinecodonsFor'D'=["GAC","GAT"]-- AspartatecodonsFor'E'=["GAA","GAG"]-- GlutamatecodonsFor'F'=["TTC","TTT"]-- PhenylalaninecodonsFor'G'=["GGA","GGC","GGG","GGT"]-- GlycinecodonsFor'H'=["CAC","CAT"]-- HistidinecodonsFor'I'=["ATA","ATC","ATT"]-- IsoleucinecodonsFor'K'=["AAA","AAG"]-- LysinecodonsFor'L'=["CTA","CTC","CTG","CTT","TTA","TTG"]-- LeucinecodonsFor'M'=["ATG"]-- MethioninecodonsFor'N'=["AAC","AAT"]-- AsparaginecodonsFor'P'=["CCA","CCC","CCG","CCT"]-- ProlinecodonsFor'Q'=["CAA","CAG"]-- GlutaminecodonsFor'R'=["AGA","AGG","CGA","CGC","CGG","CGT"]-- ArgininecodonsFor'S'=["AGC","AGT","TCA","TCC","TCG","TCT"]-- SerinecodonsFor'T'=["ACA","ACC","ACG","ACT"]-- ThreoninecodonsFor'V'=["GTA","GTC","GTG","GTT"]-- ValinecodonsFor'W'=["TGG"]-- TryptophancodonsFor'Y'=["TAC","TAT"]-- TyrosinecodonsFor'*'=["TAA","TAG","TGA"]-- StopcodonsForx=[]-- If for some reason we get bad input, treat as empty
With this element-wise function in hand, we can create a reverse translation
function to work on a protein sequence:
For each amino acid in the protein sequence this function will produce a list
of the codons that can code for it, resulting in a list of codon options. If we
wanted to ask how many different ways the protein sequence could be encoded, we
could do the following:
This folds our list up by taking the length of each sub-list and multiplying it
by the accumulated value which I initiated at 1. Note that I used genericLength
from Data.List so that it would return the length as type Integer which can
hold an arbitrarily large number (these numbers get big). Let’s give it a couple tries:
Damn, that’s a lot of ways that this short sequence (only 176 amino acids) could be
represented in DNA. Also interesting is that giving uniqueCodingsCount a sequence
with an invalid amino acid such as 'B' correctly reports that there are 0
ways to encode this protein.
Now let’s set to work creating a function that will actually generate the
unique DNA sequence combinations that can code for a protein sequence. The
signature for this function should look like this:
uniqueCodings::ProteinSeq->[DNASeq]
It’s clear that this function will make use of reverseTranslate whose result
type is [[DNACodon]], so we can begin to think how we might convert that to
[DNASeq]. To do this we can make use of a fold function (like I have already
above) with a function and an accumulator to reduce the list. So what kind of
function might we be interested in? Consider the behavior of list comprehensions:
When we draw from two lists in the creation of a new list, the list is produced
as an operation with all the combinations of the elements in the drawn lists.
To help illustrate how we can make use of this, here’s one more example:
Remember that massive number we got from uniqueCodingsCount on the lipocalin
protein sequence? We really don’t want to go printing that many of anything…
let’s keep the tests small and reasonable.
Pretty cool, the empty protein sequence can only be encoded as an empty DNA
sequence. For a lone histidine "H" we essentially get codonsFor 'H' and for
"HAM" we get a list of 9 unique combinations of codons. When I gave it
bad amino acids it tells me there is no way to encode such a sequence. We
expect length uniqueCodings s should be equal to uniqueCodingsCount s:
ghci>lettestCountss=(uniqueCodingsCounts)==(length$uniqueCodingss)ghci>testCounts""Trueghci>testCounts"MARCELINE"Trueghci>testCounts"BMO"-- Not valid protein sequenceTrue
It looks like our function checks out, we now have a way to work with the DNA
sequences that can code for a specified protein. To wrap this post up, I’ll put
that function to work solving a real world problem (I’ve done this in my own
research).
Storytime
Imagine you are a scientist who has a protein you would like to produce
and study. You know the amino acid sequence for the protein and you are
ordering the synthesis of a DNA sequence to code for your protein. For reasons
you try valiantly (but unsuccessfully) to explain to your great uncle Max during Thanksgiving
dinner, you wish to incorporate a specific DNA sequence in a particular segment
of your protein’s coding region. This is to allow a restriction enzyme to cut
your DNA sequence for important and inscrutable purposes.
Now your DNA sequence design has a constraint. If you can help it, you’d
prefer not to mutate the protein sequence. Perhaps you can take advantage of
the degeneracy of the genetic code to choose codons that provide your restriction
enzyme site without modifying any amino acids.
You select a small region of your protein that is suitable for the site: “ALGYL”
You ask, “Can I put an EcoRI or EcoRV site in here?”
The solution
You’re smart; you decide to let a computer do the investigation.
--produce only the possible coding DNA sequences that contain the constraintconstrainedUniqueCodings::ProteinSeq->DNASeq->[DNASeq]constrainedUniqueCodingssconstraint=filter(isInfixOfconstraint)$uniqueCodingss
Sadly you can’t put in an EcoRI site without changing amino acids, but you can
choose your codons to incorporate the EcoRV site. I greatly abbreviated the
output of that final evaluation, it actually gives 96 result sequences! If the
sequence search space were much larger, it could be hard to even find the codons
of note. A good way to improve the code would be to have the code identify them.
In this case the EcoRV site spans three codons for “GYL”. If we narrow the
search to just those three we get an exact notion of our options:
With that bit of practical problem solving, we conclude our demonstration.
Footnotes
1: I could employ the IUPAC compression
format to make this shorter, but that would just pass the buck and I’d have to
write the details in an IUPAC function. It would be reusable though, and might
be worth doing for that.
This post is here to provide a basic overview of the central dogma of molecular
biology as a introduction for a later post on Haskell functions pertinent to the
mechanisms in the central dogma. To get to that post, click here.
If you’re interested in a general review of the central dogma, its mechanisms
and current place in modern biology, continue reading. Feel free to skip it if
all of that is old news.
This review assumes some basic familiarity with the base-pairing nature of
nucleic acids and the structural nature of DNA, RNA, and protein.
What the “central dogma” is all about
The central dogma of molecular biology
is the codification of how information is transferred between the biopolymers:
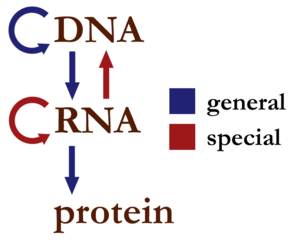
DNA, RNA, and protein. It is sometimes given in the simplest form as “DNA
proceeds to RNA proceeds to protein”. To put it in other terms, the central
dogma as a statement expresses the idea that base-sequence information
is conveyed from DNA, to RNA, ultimately to protein, and sequence information
in protein is never conveyed back to DNA or RNA. Here’s a little diagram of what
the central dogma allows:
Much has been discovered since the central dogma was originally formulated,
giving us a richer view of genetics and biology. In light of some of these
discoveries, some argue that the central dogma has been falsified, leaving it
perhaps only useful as a general statement of how most things work. I believe
that these views frequently reflect how our thinking has shifted with regards to
information in a biological context. There’s a lot more to information than
solely the sequence information with which the central dogma was originally concerned.
Falsification of the central dogma?
What are the falsification conditions of the central dogma? If any
of the following were demonstrated, we can say that the central dogma has been
demonstrated false:
Protein to Protein information flow
Protein to DNA information flow
Protein to RNA information flow
If the decoding of protein sequence to nucleic acids (reverse translation) were
to be discovered, that would be a refutation of the central dogma. So far, no
such discovery has been made. However there is some debate over whether
prion activity represents a violation.
This paper and the conversation that follows provides a good discussion of the
matter. I’ll try to give
a short layperson’s summary.
A prion is a protein
which may switch to a different folded shape and further induce others to do the
same. This is the basis of some neurodegenerative diseases as well as a form of
epigenetic heritability in some organisms. One particularly intriguing case is
that of prion activity affecting how the translation of many other proteins
takes place, the ribosome does not terminate as typical, and so affects the
sequence information of other proteins. In light of this, even keeping to a very
strict sense of the central dogma, it could be argued that protein sequence
information may result in altering protein sequence information and violate the
dogma.
I’m not sure whether or not I am fully decided here, because it
would seem to me that there is little distinction between this subject
(though certainly no less fascinating) and other epigenetic factors. I might
suggest then that if epigenetics do impinge on the central dogma, then it is
contradicted not only by prions, but other epigenetic modes as well that
influence biopolymer sequence.
Whether or not the central dogma stands does not undermine the foundations of
biology. If falsified, the central dogma may remain useful as a negation which
manages to capture a great quantity of known molecular biology. Either way, it
is vital to note that one’s understanding of biology does not end with the
central dogma.
Mechanisms of sequence information flow
Now I’ll turn the attention over to known mechanisms of information flow in the
central dogma with some examples of sequence transformations.
DNA : {A,C,G,T}
DNA to DNA occurs ubiquitously, replication of DNA by DNA
polymerases is essential for providing genetic material to an organism’s’
progeny. Fidelity of replication is important so that viable organisms produce
enough viable progeny, as is the occasional infidelity to provide diverse
progeny that might respond better to change and competition. Replication of
GATTACA would give GATTACA, with possible chance mutations.
DNA to RNA occurs ubiquitously to produce RNAs of a very wide wide variety
of functions. The process of producing RNA from DNA templates is known as
transcription and is carried out by RNA polymerases that create RNA chains in
a pair-wise complementary fashion to the DNA template. RNA is chemically very
similar to DNA, one distinguishing feature is the use of the nucleobase uracil (U)
in place of the nucleobase thymine (T) of DNA. Both are complementary to the
nucleobase adenosine (A). Thus the corresponding RNA sequence of GATTACA,
perhaps transcribed from the complementing strand would be GAUUACA.
DNA to Protein is not known to occur in nature, though it has been
accomplished in vitro under certain conditions
It is not forbidden by the central dogma, and there is speculation as to why it
is either rare or non-existant. It is important to address that we frequently
refer to the genetic code, the
coding of amino acid sequence for protein by nucleotide triplets, in DNA terms
even though it must first pass through a transcribed mRNA intermediate (more on
that below). I’ll extend GATTACA up to 9 nucleotides for this example:
GATTACAGA would translate to the amino acid sequence DYR. Be aware that the
genetic code can vary in some cases.
RNA : {A,C,G,U}
RNA to DNA occurs in many organisms and was considered a possibility from
the beginning of the central dogma, though evidence for it came somewhat later.
RNA is used as the basic genetic material for some viruses; retroviruses, HIV is
one example, must reverse transcribe their RNA genetic material to DNA before
it is integrated into the genome of the host cell. RNA templates are used in
reverse transcription to extende the telomere caps of chromosomes by
telomerases. To do reverse transcription, just reverse transcription! GAUUACA
produces GATTACA.
RNA to RNA replication occurs via RNA-dependent RNA polymerases sometimes
called replicases. This is important to the replication of viruses with RNA as
genetic material as well as eukaryotic cells for RNA silencing.
RNA Silencing is a huge deal in biology, not just for its natural role, but
because it serves as a powerful tool of basic research. Ignoring stochastic
mutations, base complementarity means that GAUUACA will give GAUUACA.
RNA to Protein is conducted by a massive complex of protein and RNA called
the ribosome, using the genetic code
that was mentioned earlier, as it scans over a messenger (mRNA) template. This
is the only known way to produce protein and is called translation.
Translation from the RNA sequence GAUUACAGA in the (not totally) universal
genetic code would produce the amino acid sequence DYR.
Protein : {A,R,D,C,Q,E,G,H,I,L,K,M,F,P,S,T,W,Y,V,...}
Protein to DNA or RNA is not known to occur in nature, in fact it is
strongly suspected to not occur. This is not permitted to occur according to
the central dogma. There’d be a Nobel Prize for whoever discovered a natural
biological system that accomplished this. It might then be interesting that this
is a common research job. Before genome sequencing was developed and became
relatively cheap, protein sequencing
might have yielded the amino acid sequence of the protein, which would need to
be reverse translated through the genetic code to either get an idea of what
the encoding gene looks like (introns and other gene features are lost data), or
to produce the protein transgenically. Protein sequencing is still an important
part of biological research, predictions made in bioinformatics on genomic
sequences need experimental validation after all. Reverse translation is made
somewhat complicated by the fact that the genetic code is degenerate, meaning
that amino acids may be coded by more than one codon, so there are many
nucleic acid sequences that could hypothetically produce the same protein.
To be brief, the choice of codon can affect how well a sequence is translated
or what other factors may regulate the mRNA; this is an area of active study.
One rudimentary approach to codon optimization is
to calculate the relative frequency of codon usage in an organism’s
protein-encoding genes and choose common codons rather than underrepresented
ones. For the sequence DYR in the universal genetic code, we would have the
following (brackets indicate choose one) [GAT,GAC][TAT,TAC][CGA,CGC,CGG,CGT,AGA,AGG]
for a total of 2 * 2 * 6 = 24 possible corresponding DNA sequences.
Protein to Protein As mentioned earlier, prions may represent a form of
protein sequence modification by other proteins and it is a matter of current
some discussion whether or not this is a refutation of the central dogma that
excludes protein to protein sequence transferrence. Protein is not replicated
from protein, nor is it produced by other means aside from translation.
On a final note
I did not mention that DNA, RNA, and protein can be modified in ways beyond
their sequence, but it is very commonly so. In addition they interact with
eachother and with various components in their biological system. Much of this
is outside the concern of the central dogma, but entirely within our concern as
scientists seeking to understand the world. So the central dogma is really a Bio
101 concept, from there we have moved on.
The wonderful and bewildering nature of biology is that it is complex. It is
the interplay of numerous agents that gives rise to the diversity of the world
in which we live, why biological research remains difficult, why some diseases
have no simple cures.
For some further reading on the complexifying factors of general information
flow in biology, the following key terms come to mind and in no particular order:
epigenetics, intron excision, alternative splicing, RNA editing, RNA silencing,
post-translational modification, inteins, riboswitches, horizontal gene
transfer.
Haskell is something I’ve always wanted to explore more deeply. I suppose that
it might be more fair to say that I really want to explore functional
programming, and Haskell currently has my attention. I believe that it holds a
lot of promise for working in bioinformatics and computational biology, for
reasons explored below.
When someone sets out to write some code to solve a problem, there’s a peristent
tradeoff decision that one generally will need to make. The following statements
are meant to be taken as painting with broad strokes. Choosing an abstract
language like Python will buy yourself reduced developer time, increased
flexibility, and more accessible code, at the expense of reduced computational
efficiency. Choosing a more “gritty” language like C will instead provide
great computational efficiency at the expense of developer time, specialization
over generalization, and less reader-friendly code.
If your problem is relatively computationally simple, you may never need to care
about computational efficiency, so you stick with the nice abstract code. If
the problem is computationally complex, you may have no other choice than to
ditch the handy abstraction and start thinking in bits and bytes. Performance
(computationally speaking) and progress (in development terms) are thus often
opposing objectives in a project. The world of programming has produced a lot of
hybrid approaches such as fast libraries of code written “close to the metal”
which can be employed by the abstract languages, or abstract languages being
used to munge data or “glue” together the pipelines of fast but inflexible
computation.
This is all well and good, and I bet it’s been summarized a gajillion times
before, so why go through all of that? Haskell is an abstract language which can
perform comparably (within an order of magnitude in many cases) to C. So perhaps
it can occupy a realm where the code can benefit from both speed of execution and
development. Haskell appears to offer decent modularity, clarity, and rapid
iterative development while also being fast and amenable to parallelization. In
terms of style, mathematical literacy translates very well to literacy in
Haskell, so computational biologists should be pretty well off.
I’ve done some fun things with Haskell; I’ve solved some Euler and Rosalind
problems, I’ve tinkered with some fun geometry, and I am literate enough now to
generally make sense of other people’s code. Yet I can’t say I’ve really done
anything particularly impressive, which is a shame. So I’ve resolved to spend
some of my free time developing my Haskell and computational biology skills, and
hope that in the future, I’ll begin a series to illustrate and share my
experiences.
This post explains how to combine use of piped input for the Libav avconv
program with a separate program generating frame data; one advantage of which is
that video may be encoded as frames are generated without accumulating (perhaps
extremely large) frame data. I’ll be illustrating it with a basic example using
molecular visualization software PyMOL though the technique is quite general.
It is my intention that this post be accessible to beginners
(PyMOL/Python/Libav).
A common workflow for those generating movies from molecular visualization
programs such as PyMOL is to script the creation of video frames in one step
followed by conversion of frames to video with Libav in a subsequent step.
One downside to this process is that keeping each individual frame on disk may
be problematic. 2 minutes worth of 1920px1080p image frames can readily weigh in
at ~3GB.
One way to work around this is to pipe the frames into avconv as they are
created and discarding them instead of accumulating them.
A worked example with dsRed
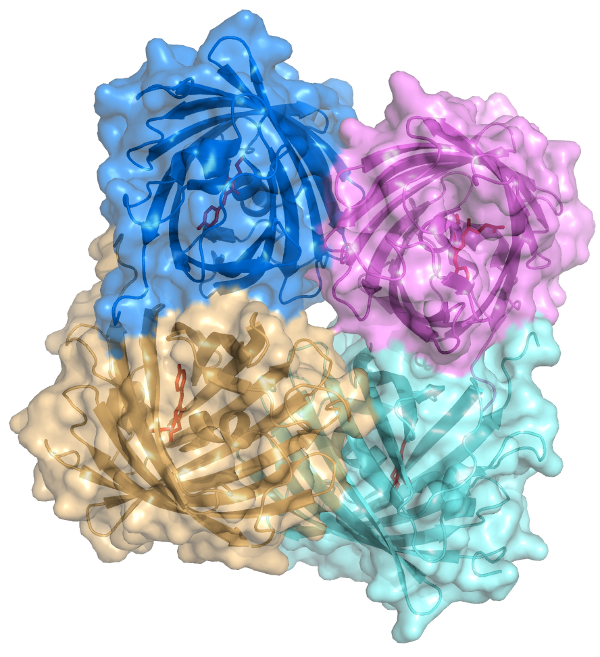
For this post’s example molecule I chose dsRed. dsRed is a coral protein which
fluoresces red in the visible spectrum. In PDB: 1G7K
we see it crystallized in its homotetrameric state.
To keep the scripts simple, I did all of my configuration in the
prepared PyMOL session. Here’s the view upon initialization:
The following script produces a small number of low resolution frames meant to
provide a pre-view of the video’s action:
frompymolimportcmddefproduce_frame():name='frame{0:03d}.png'.format(produce_frame.frame_count)cmd.png(name,ray=True,width=400,height=300)produce_frame.frame_count+=1produce_frame.frame_count=0#Rotate 90 degrees in x over 18 framesforiinrange(18):cmd.turn('x',5)produce_frame()#Rotate 360 degrees in y over 72 framesforiinrange(72):cmd.turn('y',5)produce_frame()#Rotate -90 degrees in x over 18 framesforiinrange(18):cmd.turn('x',-5)produce_frame()
We can stitch these low resolution images into a video with the following:
When ready to commit the time to a full render, the script is then updated for
appropriate number of frames and resolution:
frompymolimportcmddefproduce_frame():name='frame{0:03d}.png'.format(produce_frame.frame_count)cmd.png(name,ray=True,width=1920,height=1080)produce_frame.frame_count+=1produce_frame.frame_count=0#Rotate 90 degrees in x over 90 framesforiinrange(90):cmd.turn('x',1)produce_frame()#Rotate 360 degrees in y over 360 framesforiinrange(360):cmd.turn('y',1)produce_frame()#Rotate -90 degrees in x over 90 framesforiinrange(90):cmd.turn('x',-1)produce_frame()
Now let’s do a quick test of the piped input using our preview frame images. In
a terminal window, create a named UNIX pipe and set avconv to use this pipe as
its input, like so:
I have explicitly set the input format to image2pipe and the decoder to png,
as these things cannot be inferred in this context.
In a separate terminal window, execute the following command to send all of your
low resolution frames into the pipe on which avconv is listening.
cat *.png > my_pipe
This is the piped form of the workflow. Notes:avconv will listen until
indicated to stop or close. So if for instance we wanted to loop our video
twice with this workflow, this would do so:
cat *.png *.png > my_pipe
while this would only yield one loop:
cat *.png > my_pipe; cat *.png > my_pipe
The script is thus adapted for use with pipes.
Prior to the script’s execution, avconv must be set to
listen to the pipe;
avconv -f image2pipe -c:v png -i my_pipe dsred_high_res.mp4.
importosfrompymolimportcmddefproduce_frame():name='/tmp/frame.png'cmd.png(name,ray=True,width=1920,height=1080)withopen(name,'rb')asframe:os.write(produce_frame.pipe,frame.read())os.remove(name)produce_frame.pipe=os.open('my_pipe',os.O_WRONLY)#Rotate 90 degrees in x over 90 framesforiinrange(90):cmd.turn('x',1)produce_frame()#Rotate 360 degrees in y over 360 framesforiinrange(360):cmd.turn('y',1)produce_frame()#Rotate -90 degrees in x over 90 framesforiinrange(90):cmd.turn('x',-1)produce_frame()os.close(produce_frame.pipe)
Executing this script will result in generated frames being passed immediately
into avconv for encoding. Video encoding is also encoded simultaneously and
will conclude immediately after the frames are done being created and the pipe is
closed. Here’s the full size render.
Acknowledging some roughness
The script as presented for PyMOL results in double the file IO when adapted for
pipes. This is due to the use of the of the standard PyMOL cmd.png method. I
haven’t studied the PyMOL source enough to know how best to provide a new
pipe-friendly method for producing the PNG files, perhaps that may be a subject
for the future.